Predictors
Details on the predictors included in the database, both protein structure and disorder. Algorithm authorship is stated where necessary, as well as a contact details for all data providers.
Disorder prediction
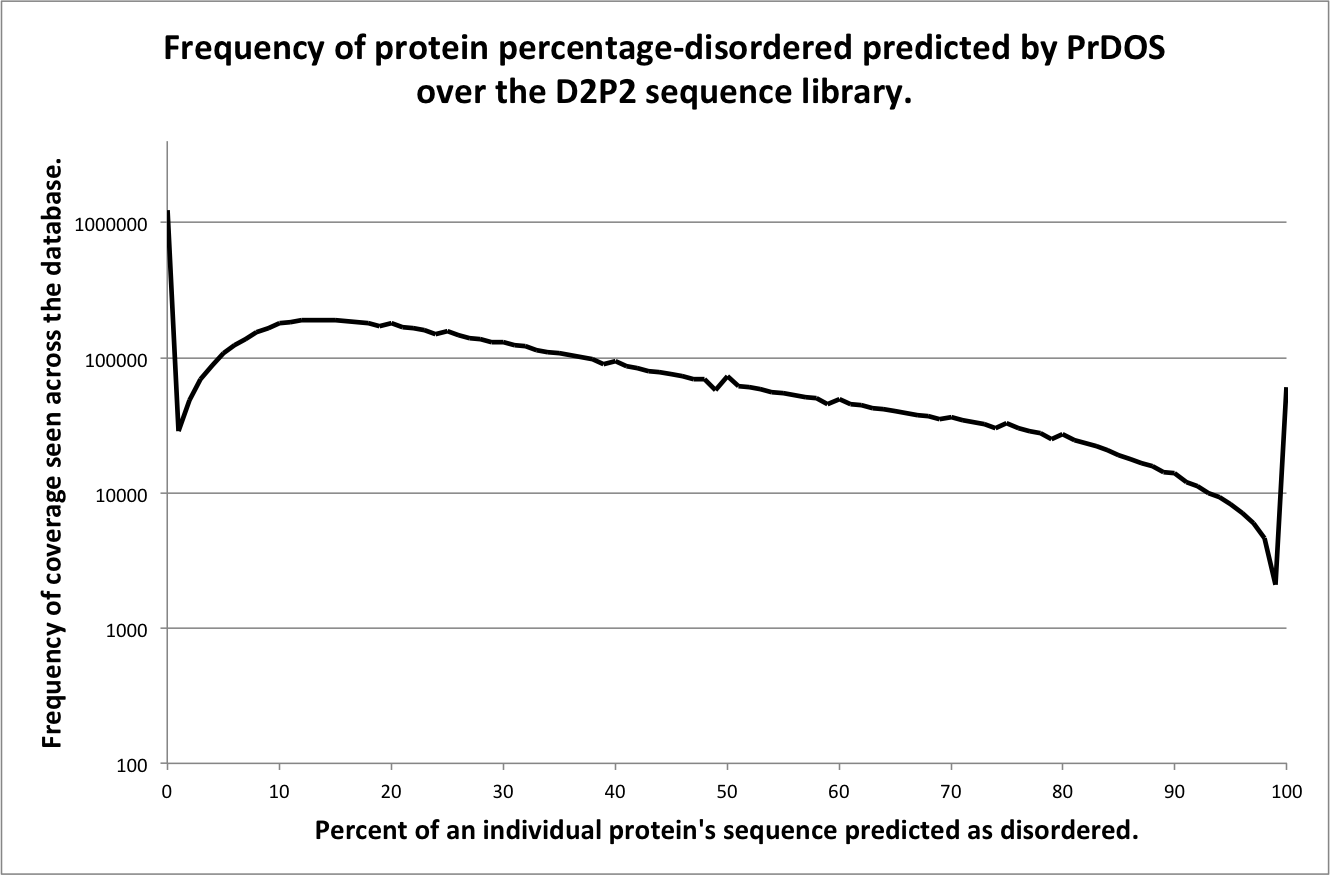
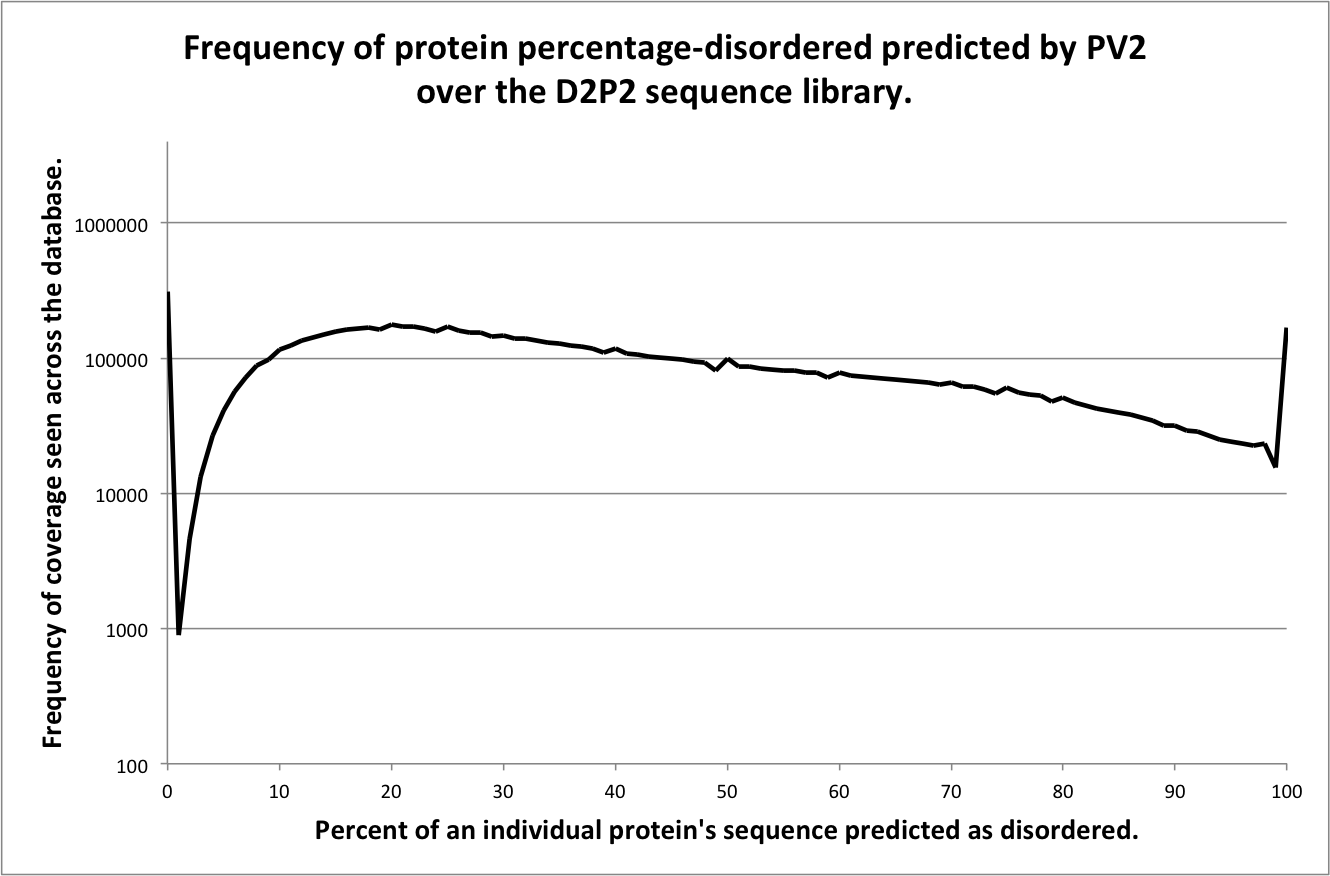
This page contains brief descriptions of the predictors included in the D2P2 assignments database, for detailed information on the methodology please refer to the cited materials. Figures included on this page show the frequency of percentage disorder coverage per protein for each predictor. The X-axis shows the percentage of a protein sequence covered with disorder prediction from a given predictor, binned at 1% intervals. The Y-axis shows the frequency of observed sequences with a given percentage coverage of disorder, log10 scaled for clarity.
If you have any specific comments or issues with a given predictors assignments a point of contact for that data provision is linked. For more general enquiries or comments please contact Matt Oates. We would also like to gratefully acknowledge the works of all original authors who were not directly involved in the construction of the D2P2 database. Finally if you are an author of a prediction method and wish to be included please get in touch we are always looking to expand the dataset.
VLXT
Description of method
PONDR® VL-XT integrates three feedforward neural networks: the VL1 predictor (Romero 2001), the N-terminus predictor (XN), and the C-terminus predictor (XC) (both from Li et al. 1999). VL1 was trained using 8 long disordered regions identified from missing electron density in X-ray crystallographic studies, and 7 long disordered regions characterised by NMR. The XN and XC predictors, together called XT, were also trained using X-ray crystallographic data, where the terminal disordered regions were 5 or more amino acids in length.
cite Romero,P., Obradovic,Z., Li,X., Garner,E.C., Brown,C.J., Dunker,A.K. (2001) "Sequence complexity of disordered protein."
Li,X., Romero,P., Rani,M., Dunker,A.K., Obradovic,Z. (1999) "Predicting Protein Disorder for N-, C-, and Internal Regions."
People involved in providing data
Any people listed are not authors of the method; unless otherwise stated in the above citation of the method.
Please direct any queries about very specific predictions and methodology to Pedro Romero who provided the source data.
- Dr Pedro Romero, Center for Computational Biology and Bioinformatics, Indiana University
- Dr Bin Xue, Department of Molecular Medicine, College of Medicine, University of South Florida
- Dr Vladimir Uversky, Department of Molecular Medicine, College of Medicine, University of South Florida
- Prof Keith Dunker, Director, Center for Computational Biology and Bioinformatics, Indiana University
 |
VSL2b
Description of method
PONDR® VSL2 predictor is a combination of neural network predictors for both short and long disordered regions (Peng 2006). A length limit of 30 residues divides short and long disordered regions. Each individual predictor is trained by the dataset containing sequences of that specific length. The final prediction is a weighted average determined by a second layer predictor (Peng et al. 2006). PONDR® VSL2 applies not only the sequence profile, but also the result of sequence alignments from PSI-BLAST and secondary structure prediction from PHD and PSIPRED. This predictor is so far the most accurate predictor in the PONDR® family.
cite Peng,K., Radivojac,P., Vucetic,S., Dunker,A.K. and Obradovic,Z. (2006) "Length-dependent prediction of protein intrinsic disorder."
People involved in providing data
Any people listed are not authors of the method; unless otherwise stated in the above citation of the method.
Please direct any queries about very specific predictions and methodology to Pedro Romero who provided the source data.
- Dr Pedro Romero, Center for Computational Biology and Bioinformatics, Indiana University
- Dr Bin Xue, Department of Molecular Medicine, College of Medicine, University of South Florida
- Dr Vladimir Uversky, Department of Molecular Medicine, College of Medicine, University of South Florida
- Prof Keith Dunker, Director, Center for Computational Biology and Bioinformatics, Indiana University
 |
PrDOS
Description of method
PrDOS is composed of two predictors. The first predictor is implemented using a support vector machine with a position-specific profile of local amino acid sequence. A similar concept to how PSIPRED predicts local secondary structure features. The second predictor assumes the conservation of intrinsic disorder in homologous protein domain families, and is implemented using PSI-BLAST and a novel measure of disorder (Ishida and Kinoshita 2007). The final prediction is taken as the combination of the results of the two predictors.
cite Ishida,T. and Kinoshita,K. (2007) "PrDOS: prediction of disordered protein regions from amino acid sequence.", Nucleic Acids Res., 35, Web Server issue
People involved in providing data
Any people listed are not authors of the method; unless otherwise stated in the above citation of the method.
Please direct any queries about very specific predictions and methodology to Takashi Ishida who provided the source data.
- Dr Takashi Ishida, Department of Computer Science, Graduate School of Information Science and Engineering, Tokyo Institute of Technology
 |
PV2
Description of method
PV2 is a meta-predictor that was built upon five prediction methodologies trained on different disordered protein datasets: logistic regression, a neural network, a support vector machine, a conditional random field, and finally PONDR® VSL2b to capture the correlation between the neighboring residues. The PV2 meta-prediction reports a residue as disordered if any two of the underlying methods agree on a disordered state (Ghalwash 2012).
cite Mohamed F. Ghalwash, A. Keith Dunker and Zoran Obradovic (2012) "Uncertainty analysis in protein disorder prediction",
People involved in providing data
Any people listed are not authors of the method; unless otherwise stated in the above citation of the method.
Please direct any queries about very specific predictions and methodology to Mohamed Ghalwash who provided the source data.
- Mr Mohamed Ghalwash, Center for Data Analytics and Biomedical Informatics, Temple University
- Prof Zoran Obradovic, Director, Center for Data Analytics and Biomedical Informatics, Temple University
- Prof Keith Dunker, Director, Center for Computational Biology and Bioinformatics, Indiana University
 |
IUPred-S
Description of method
IUPred assumes that the core of a well-structured globular protein has amino acids that can make enough favourable contacts to form a stable 3D structure. A matrix of amino acid pairs holds estimates of their pairwise interaction energies which is then used with a position specific scoring method to predict when stretches of amino acids are not contributing to a stable structure. The underlying assumption is that globular proteins are composed of amino acids which have the potential to form a large number of favourable interactions, whereas intrinsically unstructured proteins (IUPs) adopt no stable structure because their amino acid composition does not allow sufficient favourable interactions to form. (Dosztányi 2005). The S variant of IUPred uses scores specifically trained on short form disorder, and as such performs better on these regions over the long type.
cite Dosztányi,Z., Csizmók,V., Tompa,P and Simon,I. (2005) "The Pairwise Energy Content Estimated from Amino Acid Composition Discriminates between Folded and Intrinsically Unstructured Proteins"
People involved in providing data
Any people listed are not authors of the method; unless otherwise stated in the above citation of the method.
Please direct any queries about very specific predictions and methodology to Marcin Mizianty who provided the source data.
- Mr Marcin Mizianty, Department of Electrical and Computer Engineering, University of Alberta, Edmonton, Alberta, T6G 2V4, Canada
- Prof Lukasz Kurgan, Department of Electrical and Computer Engineering, University of Alberta, Edmonton, Alberta, T6G 2V4, Canada
 |
IUPred-L
Description of method
IUPred assumes that the core of a well-structured globular protein has amino acids that can make enough favourable contacts to form a stable 3D structure. A matrix of amino acid pairs holds estimates of their pairwise interaction energies which is then used with a position specific scoring method to predict when stretches of amino acids are not contributing to a stable structure. The underlying assumption is that globular proteins are composed of amino acids which have the potential to form a large number of favourable interactions, whereas intrinsically unstructured proteins (IUPs) adopt no stable structure because their amino acid composition does not allow sufficient favourable interactions to form. (Dosztányi 2005). The L variant of IUPred uses scores specifically trained on long form disorder, and as such performs better on these regions over the short type.
cite Dosztányi,Z., Csizmók,V., Tompa,P and Simon,I. (2005) "The Pairwise Energy Content Estimated from Amino Acid Composition Discriminates between Folded and Intrinsically Unstructured Proteins"
People involved in providing data
Any people listed are not authors of the method; unless otherwise stated in the above citation of the method.
Please direct any queries about very specific predictions and methodology to Marcin Mizianty who provided the source data.
- Mr Marcin Mizianty, Department of Electrical and Computer Engineering, University of Alberta, Edmonton, Alberta, T6G 2V4, Canada
- Prof Lukasz Kurgan, Department of Electrical and Computer Engineering, University of Alberta, Edmonton, Alberta, T6G 2V4, Canada
 |
Espritz-N
Description of method
Espritz predicts three variants of disorder using bi-directional recursive neural networks. Espritz-N was trained on NMR mobility data. The method can be run with a fast or slower variant (requiring PSI-BLAST) of the algorithm (Walsh 2012). Due to the wide genomic scale of D2P2 the fast variant was used. Additionally the following cut-offs were used to yield 5% false positive rate 0.3089 with this Espritz variant.
cite Walsh,I., Martin,A.J., Di Domenico,T., and Tosatto, S.C. (2012) ESpritz: accurate and fast prediction of protein disorder.
People involved in providing data
Any people listed are not authors of the method; unless otherwise stated in the above citation of the method.
Please direct any queries about very specific predictions and methodology to Marcin Mizianty who provided the source data.
- Mr Marcin Mizianty, Department of Electrical and Computer Engineering, University of Alberta, Edmonton, Alberta, T6G 2V4, Canada
- Prof Lukasz Kurgan, Department of Electrical and Computer Engineering, University of Alberta, Edmonton, Alberta, T6G 2V4, Canada
 |
Espritz-X
Description of method
Espritz predicts three variants of disorder using bi-directional recursive neural networks. Espritz-X was trained on PDB X-ray crystallography of short disorder. The method can be run with a fast or slower variant (requiring PSI-BLAST) of the algorithm (Walsh 2012). Due to the wide genomic scale of D2P2 the fast variant was used. Additionally the following cut-offs were used to yield 5% false positive rate 0.1434 for this Espritz variant.
cite Walsh,I., Martin,A.J., Di Domenico,T., and Tosatto, S.C. (2012) ESpritz: accurate and fast prediction of protein disorder.
People involved in providing data
Any people listed are not authors of the method; unless otherwise stated in the above citation of the method.
Please direct any queries about very specific predictions and methodology to Marcin Mizianty who provided the source data.
- Mr Marcin Mizianty, Department of Electrical and Computer Engineering, University of Alberta, Edmonton, Alberta, T6G 2V4, Canada
- Prof Lukasz Kurgan, Department of Electrical and Computer Engineering, University of Alberta, Edmonton, Alberta, T6G 2V4, Canada
 |
Espritz-D
Description of method
Espritz predicts three variants of disorder using bi-directional recursive neural networks. Espritz-D was trained on DisProt data for long disorder. The method can be run with a fast or slower variant (requiring PSI-BLAST) of the algorithm (Walsh 2012). Due to the wide genomic scale of D2P2 the fast variant was used. Additionally the following cut-offs were used to yield 5% false positive rate 0.5072 with this Espritz variant.
cite Walsh,I., Martin,A.J., Di Domenico,T., and Tosatto, S.C. (2012) ESpritz: accurate and fast prediction of protein disorder.
People involved in providing data
Any people listed are not authors of the method; unless otherwise stated in the above citation of the method.
Please direct any queries about very specific predictions and methodology to Marcin Mizianty who provided the source data.
- Mr Marcin Mizianty, Department of Electrical and Computer Engineering, University of Alberta, Edmonton, Alberta, T6G 2V4, Canada
- Prof Lukasz Kurgan, Department of Electrical and Computer Engineering, University of Alberta, Edmonton, Alberta, T6G 2V4, Canada
 |
Structure prediction
SUPERFAMILY
Description of method
Library of HMMs for each SCOP domain, release 1.75.
cite Gough, J., Karplus, K., Hughey, R. and Chothia, C. (2001)"Assignment of Homology to Genome Sequences using a Library of Hidden Markov Models that Represent all Proteins of Known Structure." J. Mol. Biol., 313(4), 903-919.
People involved
Authors of the method are highlighted in bold.
Please direct any queries about very specific predictions and methodology to Matt Oates who provided the source data.
- Mr Matt Oates, Intelligent Systems Lab, Department of Computer Science, University of Bristol
- Prof Julian Gough, Department of Computer Science, University of Bristol